In many areas of natural and social science, as well as engineering, data analysis involves a series of transformations: filtering, aggregating, comparing to theoretical models, culminating in the visualization and communication of results. This process is rarely static, however, and components of the analysis pipeline are frequently subject to replacement and refinement, resulting in challenges for reproducing computational results. Describing data analysis as a directed network of transformations has proven useful for translating between human intuition and computer automation. In the past I've evangelized extensively for GNU Make, which takes advantage of this graph representation to enable incremental builds and parallelization.
Snakemake is a next-generation tool based on this concept and designed specifically for bioinformatics and other complex, computationally challenging analyses. I've started using Snakemake for my own data analysis projects, and I've found it to be a consistent improvement, enabling more complex pipelines with fewer of the "hacks" that are often necessary when using Make.
I've taught Make workshops in the past, so, when I was invited to present to the Boise State University Joint User Groups, I was excited to convert that tutorial to Snakemake. Here, I've converted that tutorial into a blog post. The original (and therefore this lesson as well) is inspired by the Software Carpentry Make lesson, to which I am also a contributor.
Some prior experience with the command line is assumed, and learners are encouraged to follow along on their own computers. The entire tutorial, including questions for the learner are designed to take 2 hours as a live-coded, Software Carpentry style lesson. A standalone lesson repository can be found here and is licensed CC-BY.
Setup
If not already on your computer, install the following prerequistes.
- A Bash terminal
- Python 3.6 and the following packages
snakemakematplotlibnumpy
- The lesson assumes the following programs are also installed, but
they're not absolutely necessary for the flow of the lesson,
and/or alternatives are widely available:
headnanodottree
This example directory should be downloaded to the user's
desktop and navigated into at the command line.
(e.g. git clone https://github.com/bsmith89/zipf-example; cd zipf-example)
Motivation
Zipf's Law [10 minutes]
The most frequently-occurring word occurs approximately twice as often as the second most frequent word. This is Zipf's Law.
Let's imagine that we're interested in testing Zipf's law in some of our favorite books. We've compiled our raw data: the books we want to analyze, and have prepared several Python scripts that together make up our analysis pipeline.
The tree command produces a handy tree-diagram of the directory.
(You may not have this program installed on your computer.)
.
├── analysis.sh
├── books
│ ├── LICENSE_TEXTS.md
│ ├── abyss.txt
│ ├── isles.txt
│ ├── last.txt
│ └── sierra.txt
├── matplotlibrc
├── requirements.pip
└── scripts
├── plotcount.py
└── wordcount.py
2 directories, 10 files
Here you see that we're starting with a well designed project directory. The raw data (books) are stored in their own directory, and scripts have informative names.
Let's take a look at our raw data
head books/isles.txt
Our first step is to count the frequency of each word in a book.
scripts/wordcount.py books/isles.txt isles.tsv
Let's take a quick peek at the result.
head -5 isles.tsv
shows us the top 5 lines in the output file:
the 3822 6.7371760973
of 2460 4.33632998414
and 1723 3.03719372466
to 1479 2.60708619778
a 1308 2.30565838181
Each row shows the word itself, the number of occurrences of that word, and the number of occurrences as a percentage of the total number of words in the text file.
We can do the same thing for a different book:
scripts/wordcount.py books/abyss.txt abyss.tsv
head -5 abyss.tsv
Finally, let's visualize the results.
scripts/plotcount.py isles.tsv ascii
The ascii argument has been added so that we get a text-based
bar-plot printed to the screen.
The script is also able to render a graphical bar-plot using matplotlib and save the figure to a named file.
scripts/plotcount.py isles.tsv isles.png
Together these scripts implement a common workflow:
- Read a data file.
- Perform an analysis on this data file.
- Write the analysis results to a new file.
- Plot a graph of the analysis results.
- Save the graph as an image, so we can publish it.
Writing a script to do our analysis [5 minutes]
Running this pipeline for one book is relatively simple using the command-line. But once the number of files and the number of steps in the pipeline expands, this can turn into a lot of work. Plus, no one wants to sit and wait for a command to finish, even just for 30 seconds.
The most common solution to the tedium of data processing is to write a master script that runs the whole pipeline from start to finish.
We can see such a script in analysis.sh, which contains:
#!/usr/bin/env bash
# USAGE: bash analysis.sh
# to produce plots for isles and abyss.
scripts/wordcount.py books/isles.txt isles.tsv
scripts/wordcount.py books/abyss.txt abyss.tsv
scripts/plotcount.py isles.tsv isles.png
scripts/plotcount.py abyss.tsv abyss.png
# Archive the results.
rm -rf zipf_results
mkdir zipf_results
cp isles.tsv abyss.tsv isles.png abyss.png zipf_results/
tar -czf zipf_results.tgz zipf_results
rm -r zipf_results
This master script solved several problems in computational reproducibility:
- It explicitly documents our pipeline, making communication with colleagues (and our future selves) more efficient.
- It allows us to type a single command,
bash analysis.sh, to reproduce the full analysis. - It prevents us from repeating typos or mistakes. You might not get it right the first time, but once you fix something it'll (probably) stay that way.
What are the problems with this approach? [10 minutes]
A master script is a good start, but it has a few shortcomings.
Let's imagine that we adjusted the width of the bars in our plot
by editing scripts/plotcount.py;
in the function definition for
plot_word_counts, width = 1.0 is now width = 0.8.
Now we want to recreate our figures.
We could bash analysis.sh again.
That would work, but it could also be a big pain if counting words takes
more than a few seconds.
The word counting routine hasn't changed; we shouldn't need to recreate
those files.
Alternatively, we could manually rerun the plotting for each word-count file and recreate the archive.
for file in *.tsv; do
scripts/plotcount.py $file ${file/.tsv/.png}
done
rm -rf zipf_results
mkdir zipf_results
cp isles.tsv abyss.tsv isles.png abyss.png zipf_results/
tar -czf zipf_results.tgz zipf_results
rm -r zipf_results
But by then we've nullified many of the benefits of having a master script in the first place.
Another popular option is to comment out a subset of the lines in
analysis.sh:
#!/usr/bin/env bash
# USAGE: bash analysis.sh
# to produce plots for isles and abyss.
# These lines are commented out because they don't need to be rerun.
#scripts/wordcount.py isles.txt isles.tsv
#scripts/wordcount.py abyss.txt abyss.tsv
scripts/plotcount.py isles.tsv isles.png
scripts/plotcount.py abyss.tsv abyss.png
# Archive the results.
rm -rf zipf_results
mkdir zipf_results
cp isles.tsv abyss.tsv isles.png abyss.png zipf_results/
tar -czf zipf_results.tgz zipf_results
rm -r zipf_results
Followed by bash analysis.sh.
But this process, and subsequently undoing it, can be a hassle and source of errors in complicated pipelines.
What we really want is an executable description of our pipeline that allows software to do the tricky part for us: figuring out what steps need to be rerun. It would also be nice if this tool encourage a modular analysis and reusing instead of rewriting parts of our pipeline. As an added benefit, we'd like it all to play nice with the other mainstays of reproducible research: version control, Unix-style tools, and a variety of scripting languages.
Snakemake background [5 minutes]
Snakemake comes from a lineage of computer programs—most notably Make—originally designed to automate the compilation and installation of software. Programs like Make automate the building of target files through a series of discrete steps. Despite the original purpose, this design makes it a great fit for bioinformatics pipelines, which usually work by transforming data from one form to another (e.g. raw data → word counts → ??? → profit).
Snakemake is inspired by this approach, but designed specifically for computationally intensive and/or complex data analysis pipelines. The name is a reference to the programming language Python, which forms the basis for the Snakemake syntax. You don't need to be an expert at Python to use Snakemake, but it can sometimes be very useful. There are pros and cons to using Snakemake versus any other analysis pipeline tools, and it is worth considering other options, including:
- GNU Make
- doit
- Galaxy
Tutorial
Writing and Running Snakefiles [10 minutes]
Let's get started writing a description of our analysis for Snakemake.
Open up a file called Snakefile in your editor and add the following:
rule wordcount_isles:
input: "books/isles.txt"
output: "isles.tsv"
shell: "scripts/wordcount.py books/isles.txt isles.tsv"
We have now written the simplest, non-trivial snakefile.
The shell: line is pretty reminiscent of one of the lines from our master
script.
I bet you can already see what this snakefile means.
Let's walk through what we've written.
The first line uses the keyword rule followed by the name of our rule:
wordcount_isles.
We end that line with a colon.
All of the following lines in our rule are indented with four spaces.
The second line says that it takes an input file, using the input
keyword which is again followed by a colon.
We then give it the path to this prerequisite (books/isles.txt), wrapped in
quotes.
The third line does the same thing with the output file (isles.tsv).
And the last line is the exact shell command that we used in our shell script
earlier to create the target output file.
Like scripting, Snakemake allows us to wrap a series of shell commands, but
is more expressive and flexible than a script.
Our snakefile describes a (very short) pipeline:
- We are generating a file called
isles.tsv - Creating this file requires
books/isles.txt - The command to create this file runs the script runs
wordcount.py
We'll think about our pipeline as a network of files that are dependent on one another. Right now our Snakefile describes a pretty simple dependency graph.
books/isles.txt→isles.tsv
where the "→" is pointing from requirements to targets.
Running Snakemake
Now that we have a (currently incomplete) description of our pipeline, let's use Snakemake to execute it.
First, remove the previously generated files.
rm *.tsv *.png zipf_results.tgz
snakemake isles.tsv
You should see the following print to the terminal:
Provided cores: 1
Rules claiming more threads will be scaled down.
Job counts:
count jobs
1 wordcount_isles
1
rule wordcount_isles:
input: books/isles.txt
output: isles.tsv
jobid: 0
Finished job 0.
1 of 1 steps (100%) done
By default, Snakemake prints a summary of the recipes that it executes.
Let's see if we got what we expected.
head -5 isles.tsv
The first 5 lines of that file should look exactly like before.
Re-running Snakemake
Let's try running Snakemake the same way again.
snakemake isles.tsv
This time, instead of executing the same recipe,
Snakemake prints Nothing to be done.
What's happening here?
When you ask Snakemake to make isles.tsv it first looks at
the modification time of that target.
Next it looks at the modification time for the target's prerequisites.
If the target is newer than the prerequisites Snakemake decides that
the target is up-to-date and does not need to be remade.
Much has been said about using modification times as the cue for remaking files. This can be another Snakemake gotcha, so keep it in mind.
If you want to induce the original behavior, you only have to
change the modification time of books/isles.txt so that it is newer
than isles.tsv.
touch books/isles.txt
snakemake isles.tsv
The original behavior is restored.
Sometimes you only want Snakemake to tell you what it thinks about the
current state of your files.
snakemake --dryrun isles.tsv will print Snakemake's execution plan,
without actually carrying it out.
The flag can also be abbreviated as -n.
If you don't pass a target as an argument to snakemake (i.e. run
snakemake) it will assume that you want to build the first target in the
snakefile.
Expanding our Snakefile with more recipes (and challenge) [20 minutes]
Now that Make knows how to build isles.tsv,
we can add a rule for plotting those results.
rule plotcount_isles:
input: "isles.tsv"
output: "isles.png"
shell: "scripts/plotcount.py isles.tsv isles.png"
The dependency graph now looks like:
books/isles.txt→isles.tsv→isles.png
Let's add a few more recipes to our Snakefile.
rule wordcount_abyss:
input: "books/abyss.txt"
output: "abyss.tsv"
shell: "scripts/wordcount.py books/abyss.txt abyss.tsv"
rule archive_results:
input: "isles.tsv", "abyss.tsv", "isles.png", "abyss.png"
output: "zipf_results.tgz"
shell:
"""
rm -rf zipf_results/
mkdir zipf_results/
cp isles.tsv abyss.tsv isles.png abyss.png zipf_results/
tar -czf zipf_results.tgz zipf_results/
rm -r zipf_results/
"""
Here the recipe for zipf_results.tgz takes multiple input files,
each of which must be quoted and separated by commas, and involves
involves running a series of shell commands.
When building the archive, Snakemake will run each line successively unless
any return an error.
Question
Without doing it, what happens if you run
snakemake isles.png?Challenge
What does the dependency graph look like for your Snakefile?
Try it
What happens if you run
snakemake zipf_results.tgzright now?Practice
Write a recipe for
abyss.png.
Once you've written a recipe for abyss.png you should be able to
run snakemake zipf_results.tgz.
Let's delete all of our files and try it out.
rm abyss.* isles.*
snakemake zipf_results.tgz
You should get the something like the following output (the order may be different) to your terminal:
Provided cores: 1
Rules claiming more threads will be scaled down.
Job counts:
count jobs
1 archive_results
1 plotcount_abyss
1 plotcount_isles
1 wordcount_abyss
1 wordcount_isles
5
rule wordcount_abyss:
input: books/abyss.txt
output: abyss.tsv
jobid: 1
Finished job 1.
1 of 5 steps (20%) done
rule wordcount_isles:
input: books/abyss.txt
output: abyss.tsv
jobid: 2
Finished job 2.
2 of 5 steps (40%) done
rule plotcount_abyss:
input: abyss.tsv
output: abyss.png
jobid: 4
Finished job 4.
3 of 5 steps (60%) done
rule plotcount_isles:
input: isles.tsv
output: isles.png
jobid: 3
Finished job 3.
4 of 5 steps (80%) done
rule archive_results:
input: isles.tsv, abyss.tsv, isles.png, abyss.png
output: zipf_results.tgz
jobid: 0
Finished job 0.
5 of 5 steps (100%) done
Since you asked for zipf_results.tgz Snakemake looked first for that file.
Not finding it, Snakemake looked for its prerequisites.
Since none of those existed it remade the ones it could,
abyss.tsv and isles.tsv.
Once those were finished it was able to make abyss.png and
isles.png, before finally building zipf_results.tgz.
Try it
What happens if you
touch abyss.tsvand thensnakemake zipf_results.tgz?
Running Snakemake in parallel
And check this out!
snakemake clean
snakemake --threads 2
Did you see it?
The --threads 2 flag (just "-j2" works too) tells Make to run recipes in
two parallel threads.
Our dependency graph clearly shows that
abyss.tsv and isles.tsv are mutually independent and can
both be built at the same time.
Likewise for abyss.png and isles.png.
If you've got a bunch of independent branches in your analysis, this can
greatly speed up your build process.
Phony targets
Sometimes we want to build a bunch of different files simultaneously.
rule all:
input: "isles.png", "abyss.png"
Even though this rule doesn't have a recipe, it does have prerequisites.
Now, when you run snakemake all Snakemake will do what it needs to to bring
both of those targets up to date.
It is traditional for "all" to be the first recipe in a snakefile,
since the first recipe is what is built by default
when no other target is passed as an argument.
Another traditional target is "clean".
Add the following to your snakefile.
rule clean:
shell: "rm --force *.tsv *.png zipf_results.tgz"
Running snakemake clean will now remove all of the cruft.
Diagramming the DAG [5 minutes]
(If you'd prefer not to bake this Snakefile from scratch, you can
get one we've been hiding in the oven the whole time:
cp .extra/Snakefile.1 Snakefile)
Right now, our snakefile looks like this:
# Dummy targets
rule all:
input: "isles.png", "abyss.png"
rule clean:
shell: "rm --force *.tsv *.png zipf_results.tgz"
# Analysis
rule wordcount_isles:
input: "books/isles.txt"
output: "isles.tsv"
shell: "scripts/wordcount.py books/isles.txt isles.tsv"
rule wordcount_abyss:
input: "books/abyss.txt"
output: "abyss.tsv"
shell: "scripts/wordcount.py books/abyss.txt abyss.tsv"
# Plotting
rule plotcount_isles:
input: "isles.tsv"
output: "isles.png"
shell: "scripts/plotcount.py isles.tsv isles.png"
rule plotcount_abyss:
input: "abyss.tsv"
output: "abyss.png"
shell: "scripts/plotcount.py abyss.tsv abyss.png"
# Deliverables
rule archive_results:
input: "isles.tsv", "abyss.tsv", "isles.png", "abyss.png"
output: "zipf_results.tgz"
shell:
"""
rm -rf zipf_results/
mkdir zipf_results/
cp isles.tsv abyss.tsv isles.png abyss.png zipf_results/
tar -czf zipf_results.tgz zipf_results/
rm -r zipf_results/
"""
Looks good, don't you think?
Notice the added comments, starting with the "#" character just like in
Python, R, shell, etc.
Using these recipes, a simple call to snakemake builds all the same files
that we were originally making either manually or using the master script, but
with a few bonus features.
Now, if we change one of the inputs, we don't have to rebuild everything. Instead, Snakemake knows to only rebuild the files that, either directly or indirectly, depend on the file that changed. This is called an incremental build. It's no longer our job to track those dependencies. One fewer cognitive burden getting in the way of research progress!
In addition, a snakefile explicitly documents the inputs to and outputs from every step in the analysis. These are like informal "USAGE:" documentation for our scripts.
It is worth pointing out that our pipeline (and every pipeline) must be acyclic: no file can be an input to itself or to any of its inputs, ad infinitum. Officially we talk about the relationships between files as a Directed Acyclic Graph (DAG). While earlier we took the time to diagram our DAG by hand, Snakemake has tools for plotting this network automatically.
snakemake --dag zipf_results.tgz | dot -Tpng > dag.png
Open that file and check it out.
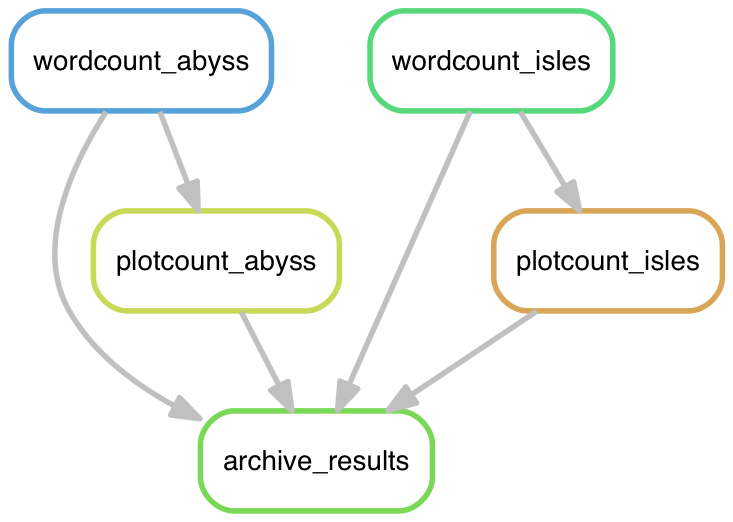
Diagrams like this one can be a very useful way to debug problems with an analysis pipeline.
Don't repeat yourself
In many programming language, the bulk of the language features are there to allow the programmer to describe long-winded computational routines as short, expressive, beautiful code. Features in Python or R like user-defined variables and functions are useful in part because they mean we don't have to write out (or think about) all of the details over and over again. This good habit of writing things out only once is known as the D.R.Y. principle.
In Snakemake a number of features are designed to minimize repetitive code. Our current snakefile does not conform to this principle, but Snakemake is perfectly capable of doing so.
Automatic variables [10 minutes]
Question
What are some of the repetitive components of our snakefile?
One overly repetitive part of our Snakefile: Inputs and outputs are in both the header and the recipe of each rule.
It turns out, that
rule wordcount_isles:
input: "books/isles.txt"
output: "isles.tsv"
shell: "scripts/wordcount.py books/isles.txt isles.tsv"
Can be rewritten as
rule wordcount_isles:
input: "books/isles.txt"
output: "isles.tsv"
shell: "scripts/wordcount.py {input} {output}"
Here we've replaced the input "books/isles.txt" in the recipe
with "{input}" and the output "isles.dat" with "{output}".
Both "{input}" and "{output}" are placeholders that refer to all of the
prerequisites and target of a rule, respectively.
In Snakemake, placeholders are all wrapped in opening and closing brackets,
and are replaced with the value of that variable at runtime.
If you are familiar with modern Python format strings, that's where the syntax
comes from.
Likewise
rule archive_results:
input: "isles.tsv", "abyss.tsv", "isles.png", "abyss.png"
output: "zipf_results.tgz"
shell:
"""
rm -rf zipf_results/
mkdir zipf_results/
cp isles.tsv abyss.tsv isles.png abyss.png zipf_results/
tar -czf zipf_results.tgz zipf_results/
rm -r zipf_results/
"""
can now be rewritten as
rule archive_results:
input: "isles.tsv", "abyss.tsv", "isles.png", "abyss.png"
output: "zipf_results.tgz"
shell:
"""
rm -rf zipf_results/
mkdir zipf_results/
cp {input} zipf_results/
tar -czf {output} zipf_results/
rm -r zipf_results/
"""
That's a little less cluttered, and still perfectly understandable once you know what the variables mean. The best part, is that if I want to change the input files, I only need to edit my snakefile in one place.
Try it
```bash snakemake clean snakemake isles.tsv ``````````
You should get the same output as last time.
Internally, Snakemake replaced "{output}" with "isles.tsv"
and "{input}" with "books/isles.txt"
before running the recipe.
Practice
Go ahead and rewrite all of the rules in Snakefile to minimize repetition and take advantage of the "
{input}" and "{output}" placeholders.
Wildcard Filenames [10 minutes]
Another deviation from D.R.Y.:
We have nearly identical recipes for abyss.tsv and isles.tsv.
It turns out we can replace both of those rules with a single rule, by telling Snakemake about the relationships between filenames with wildcards.
Using wildcards looks like this
rule wordcount:
input: "books/{name}.txt"
output: "{name}.tsv"
shell: "scripts/wordcount.py {input} {output}"
Here we've replaced the book name with "{name}".
The "{name}" matches any part of the input filename between "books/"
and ".txt", and must be the same as "{name}" in the output filename.
You don't have to use "name" as your wildcard name, and you should be
descriptive.
This rule can be interpreted as:
In order to build a file named
[something].tsv(the target) find a file namedbooks/[that same something].txt(the prerequisite) and runscripts/wordcount.py [the prerequisite] [the target].
Notice how helpful the automatic input/output variables were here. This recipe will work no matter what stem is being matched!
Go ahead and make this change in your snakefile.
Try it
After you've replaced the two rules with one rule using wildcards, try removing all of the products (
snakemake clean) and rerunning the pipeline.Is anything different now that you're using the new, universal rule?
Practice
Replace the rules for
abyss.pngandisles.pngwith a single rule.Challenge
Add
books/sierra.txtto your pipeline.(i.e.
snakemake allshould plot the word counts and add the plots tozipf_results.tgz)
(If you'd prefer a pre-cooked snakefile: cp .extra/Snakefile.2 Snakefile)
Scripts as prerequisites [10 minutes]
We've talked a lot about the power of Snakemake for rebuilding research outputs when input data changes. When doing novel data analysis, however, it's very common for our scripts to be as or more dynamic than the data.
What happens when we edit our scripts instead of changing our data?
Try it
First, run
snakemake allso your analysis is up-to-date.Let's change the default number of entries in the rank/frequency plot from 10 to 5.
(Hint: edit the function definition for
plot_word_countsinplotcount.pyto readlimit=5.)Now run
snakemake allagain. What happened?
As it stands, we have to run snakemake clean followed by snakemake all
to update our analysis with the new script.
We're missing out on the benefits of incremental analysis when our scripts
are changing too.
There must be a better way...and there is. Scripts should be considered inputs too!
Let's edit the rule for {name}.png to include plotcount.py
as an input.
rule plotcount:
input:
script="scripts/plotcount.py",
data="{name}.tsv"
output: "{name}.png"
shell: "{input.script} {input.data} {output}"
Here we've assigned names to our two inputs.
This recipe works because "{input.script}" is replaced with
"scripts/plotcount.py"
and "{input.data}" with the appropriate expansion of "{name}.tsv".
When building abyss.png, for instance,
"{input.script} {input.data} {output}" becomes
"scripts/plotcount.py abyss.tsv abyss.png", which is exactly what we want.
Try it
What happens when you run the pipeline after modifying your script again?
(Changes to your script can be simulated with
touch plotcount.py.)Practice
Update your other rules to include the relevant scripts as inputs.
(Final snakefile: cp .extra/Snakefile.3 Snakefile)
Conclusion [1 minutes]
I hope that I've convinced you of the value of Snakemake for data analysis. What I have shown you today barely scratches the surface of the software's functionality; I encourage you to check out the website. In my experience, though, the topics we've gone over today already provide 90% of the benefits: we can forget about script names and intermediate steps and focus instead on the output files that we want. This 'declarative' approach to pipelines pipelines has transformed the way I do data analysis. I think it can do the same for you.
Comments